Antibody-Drug Conjugates (ADCs) are a novel class of cancer drugs that combine three main components: a monoclonal antibody, a linker, and a toxin. By harnessing the specificity of antibodies and the potency of cytotoxic agents, ADCs can selectively target and kill tumor cells, while sparing healthy cells. The choice of toxin is critical for the efficacy and safety of ADCs, as it determines the mode and extent of cell death. Two major types of toxins are used in ADCs: microtubule inhibitors and DNA-damaging agents. Microtubule inhibitors are more prevalent, as they account for over half of the ADCs currently approved or in clinical trials. This article provides an overview of the different ADC toxins and their mechanisms of action.
1. Characteristics of ADC Payloads
ADC payloads, also known as ADC toxins or ADC effective payloads, are a crucial component of ADC drugs. As Figure 1 shows, ADC drugs enter the bloodstream and bind to target antigens on the surface of cancer cells, forming ADC-antigen complexes. These complexes then internalize into the cells. After lysosomal degradation, the complex releases the effective payload, which induces apoptosis in cancer cells. The activity and physicochemical properties of the payload directly impact the anti-tumor efficacy of ADC drugs.
More information about Antibody Internalization>>
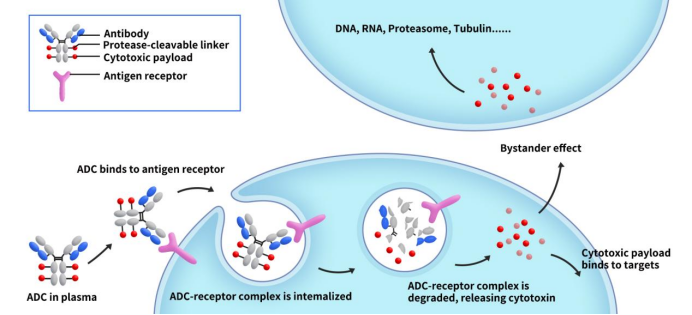
Figure 1. The mechanism od ADC action [1]
Ideal ADC toxins should have several key features, such as high cytotoxicity, low immunogenicity, high stability, modifiable functional groups or sites that do not compromise efficacy, bystander effect, suitable water solubility, and intracellular target location. These features are essential for choosing ADC payloads. Based on these criteria, we have identified the following types of ADC toxins: microtubule inhibitors, DNA synthesis inhibitors, topoisomerase inhibitors, toxic proteins, cytokines, PROTACs, and oligonucleotides.
2. Microtubule Inhibitors: Potent “Warheads” in ADC Drugs
Microtubule inhibitors are the most widely used type of “warhead” in the development of ADC drugs. Among the 15 ADC drugs currently on the market, 8 utilize microtubule inhibitors as their payloads. This category of toxins interferes with the dynamic assembly of microtubules, causing cells to arrest in the G2/M phase of the cell cycle and ultimately leading to apoptosis. Based on their different mechanisms of action, microtubule inhibitors can be further categorized into microtubule polymerization enhancers and microtubule polymerization inhibitors.
2.1 Microtubule Polymerization Enhancers
Microtubule polymerization enhancers refer to cytotoxins represented by auristatin compounds MMAE and MMAF. They typically act on the β subunit of the α,β-tubulin heterodimer, disrupting the regulated growth of microtubules. Representative examples in clinical use include Brentuximab vedotin, Polatuzumab vedotin, and Belantamab mafodotin. Learn more about MMAE/MMAF>>
2.2 Microtubule Polymerization Inhibitors
Microtubule polymerization inhibitors refer to a class of cytotoxins represented by maytansinoid compounds, with DM1 and DM4 being the primary examples. They typically disrupt the polymerization of tubulin heterodimers by inhibiting the formation of mature microtubules. Maytansinoids were first isolated from the plant Maytenus in 1972, revealing a unique macrocyclic lactam structure with nineteen members. Although maytansinoids demonstrated significant anti-proliferative effects on most cancer cell lines at subnanomolar concentrations, their lack of selectivity for cancer cells led to severe side effects in early clinical trials, preventing practical clinical applications.
It wasn’t until the early 1980s, with the emergence of the ADC concept, that researchers began to artificially modify maytansinoids, synthesizing derivatives such as DM1 and DM4. These derivatives feature a thiol modification at the C-3 position of maytansinoids. The difference lies in the group connected to the thiol moiety, with DM1 having -CH2-CH2-S- and DM4 having -CH2-CH2-C(CH3)2-S-. Currently, Kadcyla and Elahere represent the clinical applications of DM1 and DM4, respectively.
3. DNA Inhibitors
DNA inhibitors are another class of payloads for ADC drugs. Unlike microtubule inhibitors, they can damage DNA by various mechanisms, such as double-strand breaks, alkylation, intercalation, and cross-linking. They can act on any phase of the cell cycle and have potent cytotoxic effects. They have shown promising results for solid tumors [2]. Some examples of DNA inhibitors are topoisomerase I inhibitors, camptothecin, and anthracycline derivatives.
3.1 Topoisomerase I Inhibitors
Topoisomerase I inhibitors work by inhibiting the replication and transcription of DNA through their action on DNA topoisomerase I, leading to the death of tumor cells. Representative of this class is camptothecin, which forms a cleavable complex with DNA covalently. This results in a single-strand gap, with the undamaged strand rotating back into the gap once unwinding is complete. This process relaxes the supercoiled DNA, facilitating replication and transcription. After unwinding, topoisomerase I dissociates, allowing DNA chain restoration.
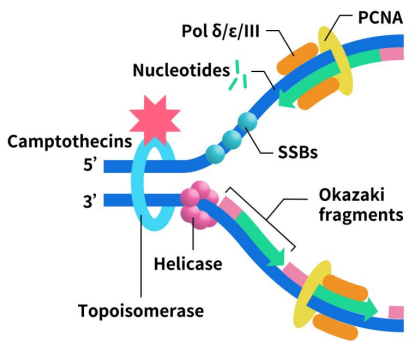
Figure 2. The mechanism of Topoisomerase I inhibitors [1]
The camptothecin derivatives commonly used in clinical applications include DXd and SN-38.
DXd, a derivative of DX-8951, is a novel, highly membrane-permeable topoisomerase I inhibitor. It exerts its anti-tumor activity by inhibiting topoisomerase I, preventing the necessary spatial changes in chromosomal DNA during the replication process, thereby blocking DNA replication and causing cell death. DXd exhibits strong anti-tumor activity, approximately ten times higher than SN-38. Its short half-life in the blood is advantageous in reducing the occurrence of toxic side effects. Additionally, DXd possesses potent cell membrane permeability, leading to bystander effects, enabling it to eliminate nearby tumor cells, and it has a shortened half-life. Among the currently marketed ADC drugs, AstraZeneca/Daiichi Sankyo’s DS-8201 specifically utilizes DXd as the active payload.
SN-38, also known as 7-ethyl-10-hydroxycamptothecin, is a semi-synthetic camptothecin. SN-38 is a metabolite of irinotecan and serves as its major anti-tumor component, with inhibitory activity 2 to 3 orders of magnitude stronger than irinotecan. However, due to its high hydrophobicity and toxicity, it can only be administered in the form of a prodrug. The hydroxyl group at position C-20 of SN-38 has the effect of reducing the intramolecular degradation rate of the lactone ring and can also be linked to the linker for ADC development. Sacituzumab govitecan, developed by Immunomedics and currently on the market, uses SN-38 as its payload.
3.2 Calicheamicins (CLM)
CLMs are a class of enediyne antibiotics isolated from rare actinomycetes, representing one of the most cytotoxic natural products. Calicheamicins primarily induce apoptosis in tumor cells by binding to specific sequences in cellular DNA grooves, leading to DNA breakage. However, they also cause damage to normal cell DNA, limiting the clinical application of calicheamicins. Despite this limitation, the high cytotoxicity, small molecular weight, and well-defined mechanism of action make calicheamicins an attractive payload for ADCs.
Among the currently marketed ADC drugs, Gemtuzumab Ozogamicin (trade name Mylotarg) and Inotuzumab Ozogamicin (trade name Besponsa) both use N-acetyl-γ1I calicheamicin as their toxin. N-acetyl-γ1I calicheamicin is a derivative of calicheamicin γ1I, with acetylation on the ethylamino sugar structure of calicheamicin γ1I. Simultaneously, to facilitate connection with amide or hydrazone linker types, its trisulfide bridge structure is modified into a disulfide bridge. Compared to other isolated analogs from fermentation broth (α2I, α3I, β1I, β1Br, γ1Br, δ1I), calicheamicin γ1I exhibits the highest cytotoxicity.
3.3 Anthracyclines
Anthracyclines are a class of antitumor antibiotics isolated by Leimgruber and others in the 1960s from the actinomycete strain Streptomyces refuineus. They belong to the Pyrrolobenzodiazepine (PBD) family. PBD compounds consist of an aromatic A-ring, a 1-4-diaza-5-1B ring, and a pyrrolidine C-ring. Their mode of action involves selective alkylation in the minor groove of DNA. The amino group at the C-2 position of the guanine in the DNA minor groove forms a covalent bond with the electrophilic iminium group inside the N-10/C-11 position of the diazepine ring, fixing the DNA’s helical structure. This process disrupts the cell division process, causing cell cycle arrest in the G2/M phase and leading to cell apoptosis.
Among the currently marketed ADC drugs, loncastuximab tesirine-lpyl (Zynlonta), developed by ADC Therapeutics, is a CD19-targeting ADC composed of a humanized IgG1κ monoclonal antibody (mAb) linked to SG3199. SG3199 is a cytotoxic PBD dimeric alkylating agent, a PBD dimer formed by connecting PBD through the C-8 position. Compared to a single PBD, the PBD dimer has a larger interaction surface with DNA, forming two covalent bonds with guanine, making the DNA fixation more robust. This property imparts increased cytotoxicity, achieving effective inhibition concentrations in the picomolar range against various tumor cells.
4. Other ADC Toxins
Pseudomonas exotoxin (PE) is the most potent toxic and immunogenic factor found in the Gram-negative bacterium Pseudomonas aeruginosa. The entire toxin consists of 638 amino acids, with the active form comprising 613 amino acids. This active form possesses ribosylation activity, capable of inactivating elongation factor-2 (EF2), thereby inhibiting protein synthesis in host cells, leading to cell death. Lumoxiti, an ADC product developed by AstraZeneca targeting CD22, features PE38 as the payload. PE38 is a truncated form of PE with reduced immunogenicity. Unfortunately, Lumoxiti was withdrawn from the market by AstraZeneca in July 2023, and the reasons for withdrawal have not been disclosed.
IRDye700DX is employed as a payload in the development of the ADC drug Akalux by Rakute Medical. It is a photosensitizing chemical substance and the world’s first photoimmunotherapy agent. After binding to the drug and cancer cells, near-infrared laser irradiation is applied to the patient. This process utilizes 690 nm near-infrared light to excite the photoactivatable chemical substance IRDye700DX (IR700) linked to the binding antibody, thereby destroying cancer cells.
5. DIMA Biotech: Empowering ADC Drug Development
DIMA Biotech is dedicated to advancing ADC research and providing researchers with the necessary tools and technical services for better-defining precision medicine. Currently, DIMA has developed multiple payload antibodies, including Anti-MMAE antibody, Anti-Dxd antibody, and Anti-SN38 antibody. These antibodies have demonstrated significant efficacy in various drug tests, holding immense potential in advancing ADC development. For other payloads, DIMA is actively arranging corresponding antibodies. If you have any development suggestions, please feel free to leave us a message.
| Payload target | Product Name | Cat. No. |
| MMAE | Anti-MMAE antibody(11C8); Rabbit mAb | DME101007 |
| Anti-MMAE antibody(11B2); Rabbit mAb | DME101006 | |
| Anti-MMAE antibody(8B4); Rabbit mAb | DME101003 | |
| Anti-MMAE antibody(8C4); Rabbit mAb | DME101004 | |
| Anti-MMAE antibody(9C4); Rabbit mAb | DME101005 | |
| Dxd | Anti-Dxd antibody(1A12); Rabbit mAb | DME101026 |
| Anti-Dxd antibody(1A1); Rabbit mAb | DME101024 | |
| Anti-Dxd antibody(1A5); Rabbit mAb | DME101025 | |
| Anti-Dxd antibody(1E6); Rabbit mAb | DME101027 | |
| SN38 | Anti-SN38 antibody(1G1); Rabbit mAb | DME101020 |
References:
[1] Wang Z, Li H, Gou L, Li W, Wang Y. Antibody-drug conjugates: Recent advances in payloads. Acta Pharm Sin B. 2023 Oct;13(10):4025-4059.
[2] Amani J., Gorjizadeh N., Younesi S., Najafi M., Ashrafi A.M., Irian S., et al. Cyclin-dependent kinase inhibitors (CDKIs) and the DNA damage response: the link between signaling pathways and cancer. DNA Repair. 2021;102.