In April 2025, a clinical case report in Nature Medicine offered groundbreaking evidence for precision therapeutics targeting SCN2A. The study described a neonate with early-onset, drug-resistant epilepsy caused by a gain-of-function (GoF) SCN2A mutation—encoding the α-subunit of the voltage-gated sodium channel NaV1.2. Following intrathecal administration of elsunersen, an SCN2A-targeting antisense oligonucleotide (ASO), the patient experienced a marked reduction in seizure frequency and showed early neurological improvement. Although this was a single case, it provided the first human proof that modulating SCN2A expression can directly alter disease trajectory in SCN2A-related disorders [1].
In parallel, early-phase trials of small-molecule SCN2A modulators have also reported encouraging signals [2], suggesting that genetic neurological diseases once considered untreatable may now be addressed through targeted intervention. Together, these developments highlight both therapeutic opportunity and scientific urgency: defining the molecular mechanisms, functional consequences, and clinical spectrum of SCN2A is essential for advancing precision medicine strategies.
1. SCN2A / NaV1.2 Structure
SCN2A, located on human chromosome 2q24.3, encodes the NaV1.2 α-subunit, a core component of voltage-gated sodium channels (VGSCs). VGSCs are essential ion channels in the nervous system, where their rapid opening and closing mediate the initiation and propagation of action potentials.
As the Figure 1A shows, NaV1.2 consists of four homologous domains (DI–DIV), each containing six transmembrane helices (S1–S6). The S4 helix functions as the voltage sensor, while the S5–S6 helices and connecting loops form the ion-conducting channel pore. The DEKA amino acid motif within the pore acts as the selectivity filter, determining sodium ion permeability and selectivity [3]. The cytoplasmic loop connecting repeats III and IV forms the fast inactivation gate, critical for controlling action potential duration.
Currently, two isoforms of NaV1.2 have been identified, differing at amino acid position 209. NaV1.2 interacts with AnkyrinG (ANK3) to anchor the channel to the neuronal membrane and binds calmodulin, modulating channel function. Stop codons before the nonsense-mediated decay (NMD) boundary can terminate protein translation, whereas stop codons beyond this boundary may not inhibit translation.
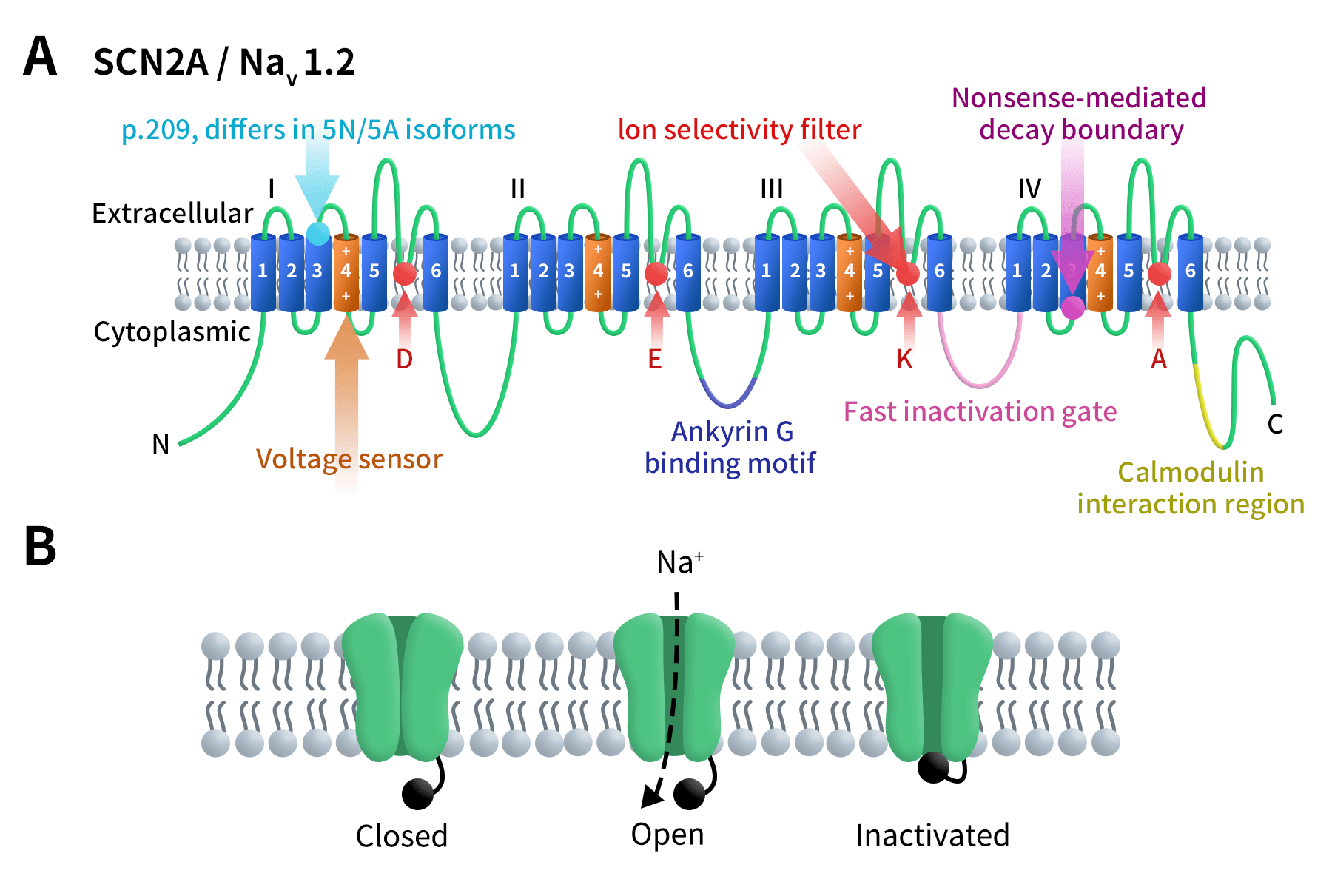
Figure1. The structure of SCN2A/NaV1.2 [4]
As the Figure 1B shows, at the resting membrane potential, NaV1.2 remains closed. Depolarization opens the channel, allowing sodium ion influx, which is subsequently blocked by the fast inactivation gate. Upon return to the resting state, the channel resets, ready for the next action potential.
2.Sodium Channel Family and SCN2A/NaV1.2
As mentioned above, voltage-gated sodium channels (VGSCs, NaV) are essential proteins mediating neuronal excitatory signal transmission in the central and peripheral nervous systems. VGSCs are composed of a pore-forming α-subunit and auxiliary β-subunits. In humans, NaV α-subunits are encoded by SCN1A–SCN11A (NaV1.1–NaV1.9), with each isoform showing distinct expression patterns and functional roles in neural development, action potential initiation, and neuronal network activity [3]. Among these, NaV1.1, NaV1.2, NaV1.3, and NaV1.6 are critically involved in CNS development and epilepsy pathogenesis.
Within the central nervous system, NaV1.1 (SCN1A) is primarily expressed in inhibitory neurons, regulating inhibitory signaling. In contrast, NaV1.2 (SCN2A) is highly localized to the axon initial segment (AIS) and dendrites of excitatory neurons, where it governs action potential initiation and signal propagation [5]. During postnatal development, NaV1.6 (SCN8A) gradually replaces part of NaV1.2 function; however, NaV1.2 expression in dendrites and unmyelinated axons remains indispensable. This spatiotemporal specificity of SCN2A explains why SCN2A mutations can lead to diverse clinical phenotypes depending on the developmental stage, including epilepsy, autism spectrum disorder (ASD), or cognitive impairment [5].
3.Mechanisms of SCN2A/NaV1.2 Function
NaV1.2, the α-subunit of voltage-gated sodium channels, is a key excitable membrane channel in CNS excitatory neurons. It is highly enriched in the axon initial segment (AIS) and dendrites, where it supports neuronal excitability and drives action potential initiation and propagation. Under physiological conditions, NaV1.2 tightly regulates sodium influx through rapid activation and inactivation, ensuring timely action potential firing and stable network activity.
Pathogenic SCN2A variants disrupt this process, impairing neuronal firing, synaptic transmission, and circuit development. Functionally, these variants fall into gain-of-function (GoF) or loss-of-function (LoF) categories. GoF variants increase activation or hinder inactivation, causing neuronal hyperexcitability typical of neonatal and infantile epilepsy. LoF variants reduce channel expression or sodium current density, leading to hypoexcitability associated with intellectual disability (ID), ASD, and related neurodevelopmental disorders [6].
However, emerging evidence shows that SCN2A dysfunction extends beyond this simple GoF–LoF framework. Human iPSC-derived neuron studies reveal mixed or asymmetric deficits, where unstable variants are prematurely degraded, markedly reducing NaV1.2 protein levels and impairing sodium currents and action potential firing [7]. Additionally, certain SCN2A mutations correlate with malformations of cortical development (MCDs), indicating that disrupted neuronal maturation and cortical network formation also contribute to altered cortical architecture and neurodevelopmental outcomes beside ion channel dysfunction [8].
4.Advances in SCN2A/NaV1.2 Drug Research and Precision Therapies
The pathogenic mechanisms of SCN2A are multifaceted and highly relevant to neurodevelopmental disorders and epilepsy. These mechanisms include alterations in NaV1.2 channel biophysics (such as changes in activation or inactivation gating), decreased channel expression or stability, abnormal neuronal development and circuit formation, and complex gene-environment and developmental timing interactions. Together, these mechanisms determine the clinical spectrum of SCN2A-related disorders, ranging from neonatal epilepsy and developmental epileptic encephalopathy (DEE) to presentations limited to ASD or ID.
Current treatment strategies for SCN2A-associated disorders are rapidly evolving along three major pathways in precision medicine and targeted therapy:
- Precision small molecules, designed to modulate NaV1.2 sodium channel activity and correct GoF or abnormal activation.
- ASOs and individualized oligonucleotides, aimed at reducing expression of the pathogenic SCN2A allele (primarily for GoF variants) or restoring expression (strategies for LoF variants are still in early exploration).
- Gene therapy, AAV, and gene-editing approaches, targeting functional restoration or replacement of NaV1.2, mostly in preclinical or early development stages.
Clinical and preclinical evidence for these approaches is accumulating, with some drugs already in multinational clinical trials or receiving regulatory incentives. According to current statistics, there are 26 drugs worldwide targeting SCN2A, including 13 in preclinical development, 9 discontinued or showing no progress, and only 4 in active clinical development.
4.1 Small Molecule Compounds Targeting SCN2A/NaV1.2
Tetrodotoxin (TTX) is a highly selective voltage-gated sodium channel blocker, targeting SCN1A, SCN2A, and SCN5A, developed by WEX Pharmaceuticals. TTX rapidly suppresses neuronal action potential firing and is primarily explored for analgesia and neuropathic pain. Its key candidate, Halneuron (TTX injection), has entered Phase III clinical trials for cancer pain, chemotherapy-induced neuropathy (CINP), and other chronic refractory pain conditions. Halneuron has received FDA Fast Track designation, and the company completed regional licensing agreements in China. As a non-opioid analgesic with a novel mechanism, TTX remains in late-stage clinical validation and is not yet approved for market.
NBI-355, developed by Neurocrine Biosciences, is a small-molecule sodium channel blocker targeting NaV1.2/NaV1.6 (encoded by SCN2A/SCN8A), intended for epilepsy therapy. Its mechanism involves blocking hyperactive sodium channels, reducing pathological neuronal firing. NBI-355 is currently in Phase I clinical trials.
4.2 Antisense Oligonucleotides (ASOs) Targeting SCN2A/NaV1.2
Elsunersen (PRAX-222), developed by Praxis Precision Medicines with Ionis Pharmaceuticals and RogCon, Inc., is an ASO that selectively reduces SCN2A mRNA expression, decreasing NaV1.2 production and correcting neuronal hyperexcitability caused by GoF mutations. Currently, Elsunersen is in Phase III registration trials for early-onset SCN2A-DEE and has received FDA Orphan Drug Designation (ODD), Rare Pediatric Disease (RPD) designation, and EMA PRIME eligibility.
nL-SCN2A-002, developed by n-Lorem Foundation, is an individualized ASO that selectively induces RNase H-mediated degradation of mutant SCN2A transcripts, aiming to reduce pathogenic NaV1.2 expression and alleviate GoF epilepsy. This candidate is advanced in N-of-1 Phase I/II trials via intrathecal administration (clinical registry NCT06314490). Early case reports indicate good safety and preliminary evidence of reduced seizure frequency. It remains in early clinical/researcher-initiated trials.
Additionally, several preclinical SCN2A-targeting therapies are under investigation, including small molecules, synthetic peptides, and AAV-based gene therapies, highlighting the broad pipeline for precision medicine in SCN2A-related disorders.
5.DIMA BIOTECH’s SCN2A Nanodisc Full-Length Membrane Protein Supports NaV1.2-Targeted Drug Development
Although SCN2A-targeted drug development now includes multiple approaches such as ASOs and small molecules, all mechanistic studies and screening efforts fundamentally require high-quality NaV1.2 membrane protein. The full-length voltage-gated sodium channel is highly complex, with 24 times transmembrane domains, and traditional soluble fragments or crude membrane preparations often cannot support structural analysis, binding-site validation, or compound screening. Therefore, nanodisc-reconstituted full-length SCN2A, which preserves native channel conformation, has become an essential tool for mechanistic research and lead optimization.
Human SCN2A full length protein-synthetic nanodisc (Cat.No.FLP100723)
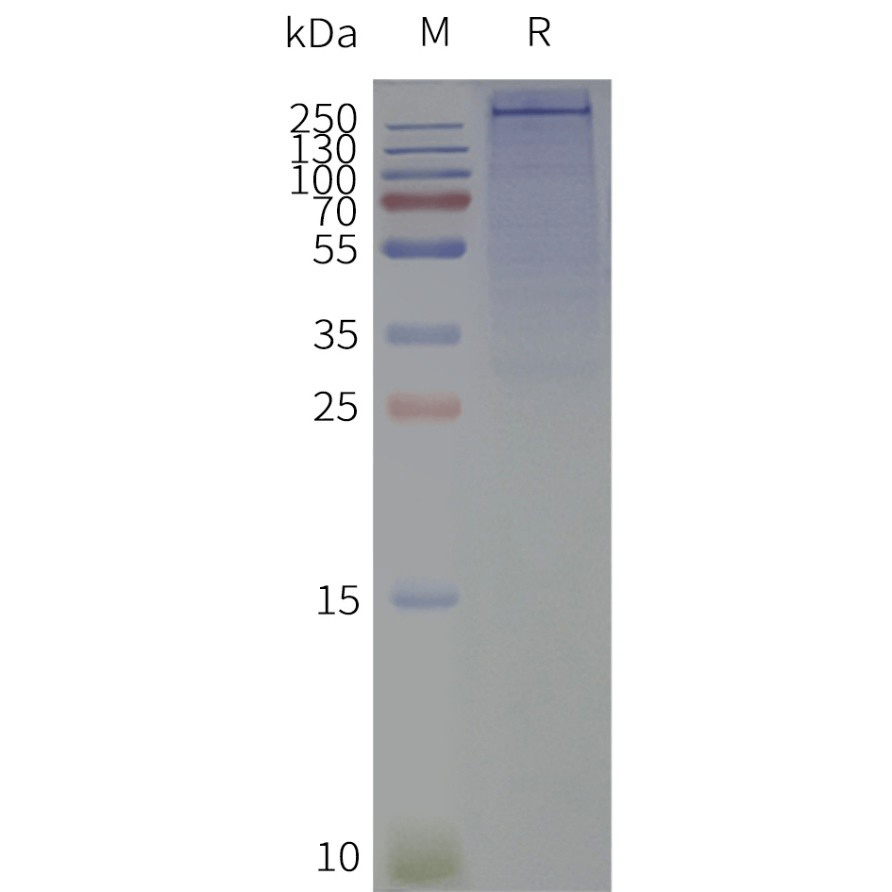
Human SCN2A-Nanodisc, Flag Tag on SDS-PAGE
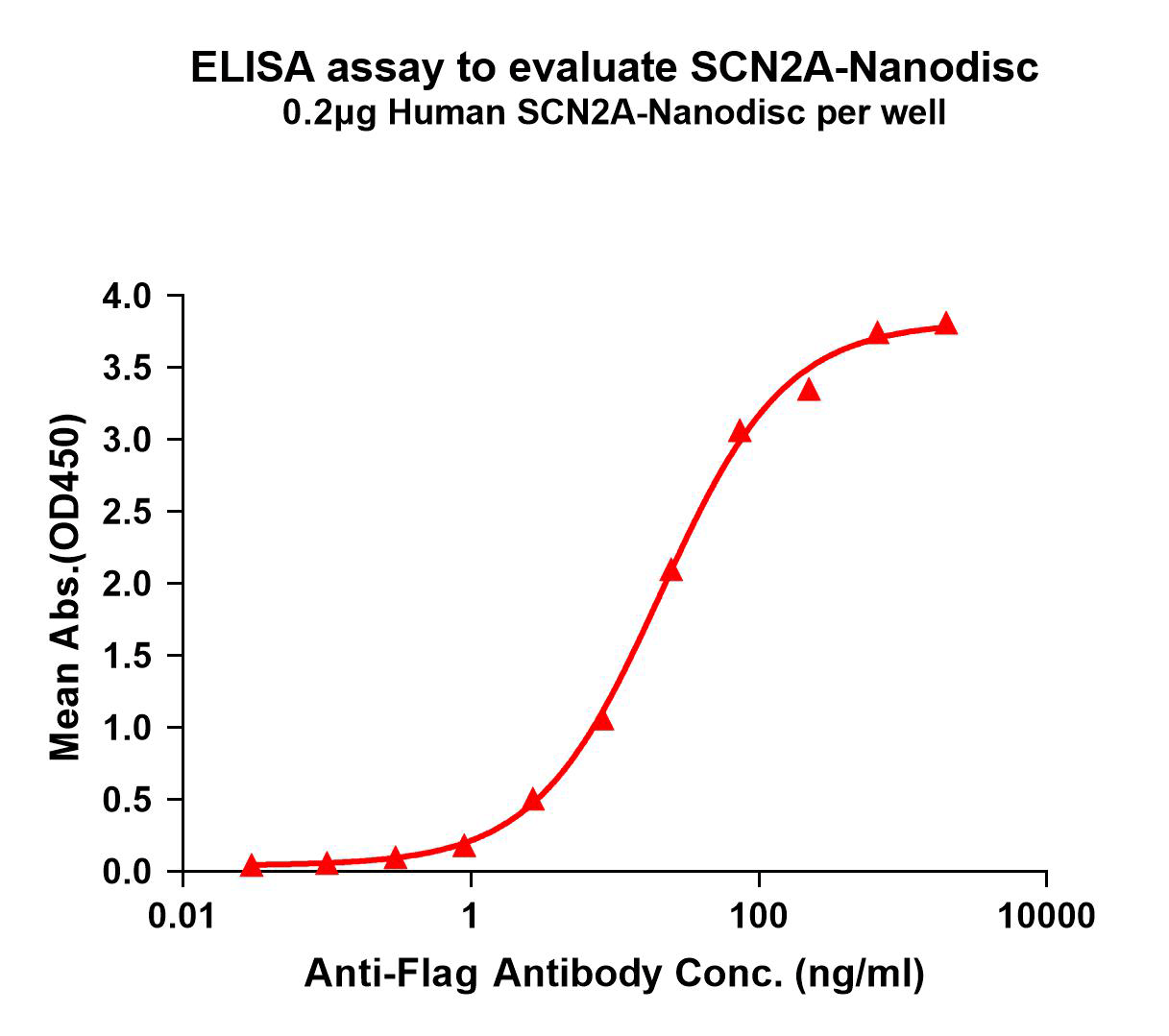
SCN2A-Nanodisc binding with anti-Flag mAb
In addition, as a professional membrane protein manufacturer, Dimabio can also provide other proteins from the same family for your cross-screening studies.
|
Target |
Cat.No. |
Product Name |
|
SCN3A/Nav1.3 |
FLP120724 |
|
|
SCN4A/Nav1.4 |
FLP120725 |
|
|
SCN5A/Nav1.5 |
FLP100726 |
|
|
SCN8A/Nav1.6 |
FLP120727 |
Human SCN8A-Strep full length protein-synthetic nanodisc IN STOCK |
|
SCN9A/Nav1.7 |
FLP120728 |
Reference:
- Wagner, M., Berecki, G., Fazeli, W. et al. Antisenseoligonucleotide treatment in a preterm infant with early-onset SCN2A developmental and epilepticencephalopathy. Nat Med 31, 2174–2178 (2025).
- [2] ClinicalTrials. gov. NCT05737784 PRAX-222 in Pediatric Participants With Early-Onset SCN2A-DEE (EMBRAVE).
- [3] Thompson CH, et al. Epilepsy-associated SCN2A (NaV1.2) variants exhibit diverse and complex functional properties. J Gen Physiol. 2023.
- [4] Sanders SJ, Campbell AJ, Cottrell JR, etal. Progress in Under standing and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018 Jul; 41(7):442-456.
- [5] Sanders SJ. Progress in Understanding and Treating SCN2A-Mediated Disorders. 2018.
- [6]Zeng Q., et al. SCN2A-Related Epilepsy: The Phenotypic Spectrum, Treatment and Prognosis. Frontiers in Molecular Neuroscience. 2022.
- [7] RA sadollahi, IDelvendahl, RMuff, et al, Pathogenic SCN2A variants causeearly-stage dysfunction in patient-derived neurons, Human Molecular Genetics, Volume 32, Issue 13, 1July 2023, Pages 2192–2204.
- [8] ClatotJ, Thompson CH, Sotardi S, et al. Rare dysfunctional SCN2A variants are associated with malformation of cortical development. Epilepsia. 2025 Mar; 66 (3): 914-928.