In the competitive landscape of cancer treatment, “ferroptosis” is emerging as a breakthrough approach to combat drug-resistant tumors. While malignant tumors maintain high proliferation through metabolic remodeling, they also bear a heavy oxidative stress burden. This inherent metabolic vulnerability forces tumor cells to rely heavily on a complex defense system, with Solute Carrier Family 7 Member 11 (SLC7A11), also known as xCT, being a crucial guardian on this front. As the core subunit of the System xc- (a heterodimeric amino acid transport protein complex), SLC7A11 mediates cysteine uptake, a rate-limiting step in the synthesis of the antioxidant glutathione (GSH), directly determining the tumor’s ability to resist ferroptosis. Research published in early 2025 in top journals such as Cell and Nature highlighted that SLC7A11 not only sustains tumor survival but may, under certain conditions, trigger disulfidptosis (a form of cell death) [1][2]. This shift from a protective role to an antagonistic one has positioned SLC7A11 as one of the most promising broad-spectrum anticancer targets. This article explores the protein structure of SLC7A11, delves into its regulatory mechanisms in ferroptosis, and reviews the latest clinical research advancements.
1. SLC Protein Family and SLC7A11
SLC7A11 is an important amino acid transporter in the human body and belongs to the SLC7 subfamily of the solute carrier (SLC) protein family. The SLC protein family is one of the largest membrane transport protein families, comprising over 400 transporters across more than 60 subfamilies. These proteins are involved in the transmembrane transport of amino acids, glucose, nucleotides, ions, and metabolites. Key members include glucose transporters of the SLC2 family (e.g., GLUT1, GLUT4), neutral amino acid transporters of the SLC1 family (e.g., SLC1A5), cationic amino acid transporters of the SLC7 family (e.g., SLC7A11), and lactate transporters of the SLC16 family. These transporters play vital roles in metabolic regulation, neuronal signaling, tumor nutrient dependence, and drug uptake, making them important sources for the development of metabolic and cancer drugs.
In the SLC7 family, based on structural and functional differences, members are classified into cationic amino acid transporters (CATs) and light chain amino acid transporters (LATs). SLC7A11 belongs to the LATs category and is a typical multi-pass transmembrane protein consisting of about 501 amino acids and containing 12 transmembrane helices. Both its N-terminal and C-terminal are located on the cytosolic side, with the central transmembrane region forming a substrate-binding and exchange channel.
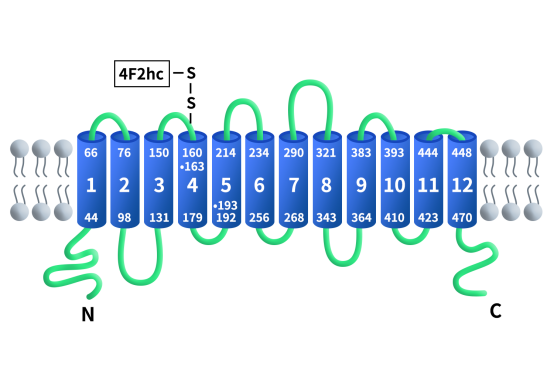
Figure 1. The structure of SLC7A11/xCT [3]
2. Distribution and Function of SLC7A11
Under normal physiological conditions, SLC7A11 is relatively highly expressed in brain tissues (microglial cells, astrocytes) and immune cells. However, in various malignancies (such as non-small cell lung cancer, pancreatic cancer, glioma, and breast cancer), driven by transcription factors like NRF2, SLC7A11 is highly overexpressed, becoming a protective shield for tumor cells against oxidative stress. As previously mentioned, SLC7A11 is a functional subunit of the System xc- reverse transport system. This system consists of the light chain SLC7A11 and the heavy chain SLC3A2 (CD98hc), which form a heterodimer linked by a disulfide bond and are co-localized on the cell membrane. The disulfide bond is formed between the conserved cysteine residue at position 158 of SLC7A11 and the cysteine residue at position 109 of SLC3A2. SLC3A2 is a single-pass transmembrane protein that acts as a molecular chaperone, maintaining the stability and proper membrane localization of SLC7A11, while SLC7A11 is responsible for substrate specificity recognition and transport function.
SLC7A11 exchanges extracellular cysteine for intracellular glutamate on a 1:1 molar basis, providing cysteine for GSH synthesis and maintaining cellular redox homeostasis. As shown in the figure, extracellular cysteine enters the cell through SLC7A11 and is then reduced to cysteine via an NADPH-consuming reaction. Cysteine is then used in a two-step process to synthesize GSH. First, cysteine combines with glutamate under the catalysis of γ-glutamylcysteine synthetase (γ-GCS) to form γ-glutamylcysteine. In the second step, the enzyme-mediated reaction by glutathione synthetase (GS) adds glycine to γ-glutamylcysteine to form GSH. Over-accumulation of lipid peroxides in the cell membrane induces ferroptosis. Glutathione peroxidase 4 (GPX4) uses GSH to reduce lipid peroxides into lipid alcohols, thus inhibiting ferroptosis. In this process, GSH is oxidized to GSSG, which is then reduced back to GSH by glutathione reductase (GR) via an NADPH-consuming reaction.
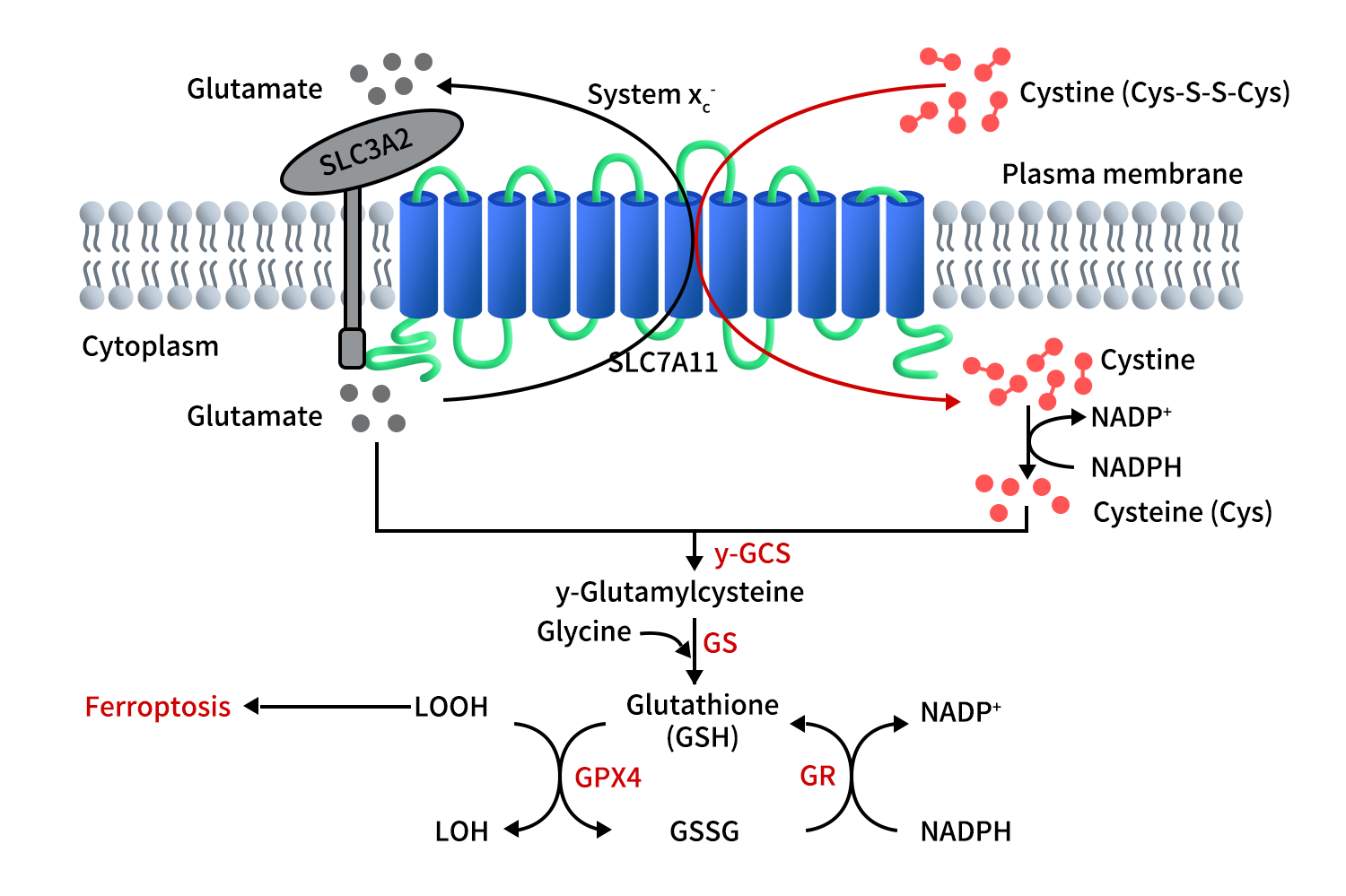
Figure 2. The function of SLC7A11 [3]
3. Mechanisms of SLC7A11 in Tumor Cells
Tumor cells often exist in a state of high oxidative stress and high metabolic activity, heavily relying on the antioxidant system. Thus, SLC7A11 plays a critical role in tumor survival and proliferation by maintaining GSH levels.
In ferroptosis regulation, SLC7A11 is a key upstream node. GSH is a cofactor for GPX4, which reduces lipid peroxides to prevent the accumulation of membrane lipid peroxides. When SLC7A11 is inhibited or downregulated, GSH depletion, reduced GPX4 activity, and accumulation of lipid ROS trigger ferroptosis. Therefore, SLC7A11 is considered one of the determinants of ferroptosis sensitivity and is a crucial intervention target in recent tumor metabolism therapy.
From a signaling regulation perspective, SLC7A11 is regulated by multiple tumor-associated pathways. The tumor suppressor gene p53 can transcriptionally inhibit SLC7A11 expression, increasing the cell’s sensitivity to ferroptosis. On the other hand, NRF2 can upregulate SLC7A11 transcription under oxidative stress, promoting antioxidant defense. Furthermore, metabolic-related transcription factors such as ATF4 and MYC can also enhance its expression, helping tumor cells maintain a reducing environment under nutrient scarcity or treatment stress. This “metabolic dependency oxidative protection” axis makes SLC7A11 a significant molecular basis for tumor resistance, such as in chemotherapy, radiotherapy, and some targeted therapies, where SLC7A11 upregulation can reduce ROS damage induced by treatment. Additionally, SLC7A11’s role in glutamate efflux may alter the tumor microenvironment, affecting immune cells and neuro-signaling pathways. In gliomas and other cancers, excessive glutamate release is associated with enhanced tumor invasiveness [3][4][5][6].
In recent research by Professor Gan Peixue and others in 2023 and 2024, an interesting phenomenon was revealed: while SLC7A11 protects against ferroptosis, under glucose deprivation, extremely high levels of SLC7A11 can lead to excessive accumulation of intracellular cysteine and other disulfides. Due to the lack of reducing equivalents (NADPH), the surplus disulfides cause collapse of the actin cytoskeleton, inducing a unique form of cell death known as disulfidptosis. This discovery provides theoretical support for developing a “SLC7A11 high-expression + metabolic inhibition” combination therapy [7][8].
4. Clinical Research Progress on SLC7A11 Drugs
Abnormal expression of SLC7A11 is closely associated with various diseases, particularly in tumors where it is significantly upregulated, such as pancreatic cancer, triple-negative breast cancer, lung cancer, hepatocellular carcinoma, and glioma, often correlating with tumor progression and poor prognosis. SLC7A11 is also involved in regulating oxidative stress in diseases like Alzheimer’s and ischemia-reperfusion injury. Given its central role in redox imbalance and metabolic reprogramming, SLC7A11 has become an important therapeutic target. Despite its importance, there is still a lack of highly selective drugs available. Current strategies can be categorized as follows:
4.1 Traditional Small Molecule Inhibitors
- Sulfasalazine, initially developed and marketed by Pfizer for the treatment of rheumatoid arthritis and inflammatory bowel disease, was later found to inhibit SLC7A11-mediated cysteine uptake, thereby inducing ferroptosis. This drug is the only SLC7A11 inhibitor currently in clinical evaluation, but due to poor pharmacokinetics and low selectivity, it has shown limited results in clinical trials.
- Sorafenib, developed by Bayer, is a marketed multi-kinase inhibitor used in hepatocellular carcinoma and renal cancer treatment. Subsequent mechanistic studies found that its anticancer activity may partially involve the regulation of ferroptosis pathways, indirectly affecting the SLC7A11-related metabolic axis. However, Sorafenib is not a selective SLC7A11 inhibitor, and its ferroptosis effects are more of a mechanistic discovery than a targeted therapeutic development.
4.2 Next-Generation Highly Selective Small Molecules
- FC-001 is one of the few selective SLC7A11 inhibitors globally that has progressed into clinical trials. Developed by the Japanese biotechnology company, FerroptoCure, this drug has reached Phase I clinical trials and is primarily targeting advanced solid tumors to assess its safety and preliminary efficacy. FC-001 is regarded as a potential “first-in-class” ferroptosis-targeting molecule and is still in the early stages of clinical validation.
- Erastin, along with its modified derivatives like Imidazole Ketone Erastin (IKE), was initially discovered by academic teams from Columbia University. Erastin is a well-known ferroptosis inducer that directly inhibits the functionality of SLC7A11. However, due to its poor in vivo stability and solubility, it has not advanced to clinical trials. Subsequent developments, such as IKE, aim to optimize the pharmacokinetic properties of Erastin, but these compounds are still in preclinical stages.
In addition to traditional small molecule inhibitors, emerging therapeutic strategies targeting SLC7A11 are gradually being developed. Notably, PROTAC (Proteolysis Targeting Chimeras) and tumor vaccine approaches are gaining attention, reflecting a shift from functional inhibition to protein degradation and immune neutralization. These approaches, however, remain in the early stages of research and have yet to produce significant breakthroughs.
5.DIMA BIOTECH’s SLC7A11 Nanodisc: Accelerating Drug Discovery for SLC7A11
Despite the availability of various strategies for SLC7A11 drug development, including small molecules and vaccines, all mechanistic research and drug screening rely heavily on high-quality SLC7A11 membrane proteins. The full-length SLC7A11 protein is complex, with many transmembrane domains, making it difficult for traditional soluble fragments or crude cell membrane preparations to meet the structural analysis, binding site validation, and drug screening needs.
Thus, SLC7A11 nanodisc full-length membrane proteins, which stably present the protein in its natural conformation, are becoming essential tools for mechanistic research and lead compound evaluation. DIMA BIOTECH has successfully utilized its mammalian cell expression system to prepare full-length SLC7A11 proteins with native conformation and intact transmembrane structure. These proteins maintain their natural conformation and post-translational modifications in detergent-free environments, providing a more reliable target model for structural analysis, antibody screening, and drug-binding tests. This technology is gradually becoming an important resource in early-stage drug development.
Human SLC7A11 full length protein-synthetic nanodisc (FLP100048)
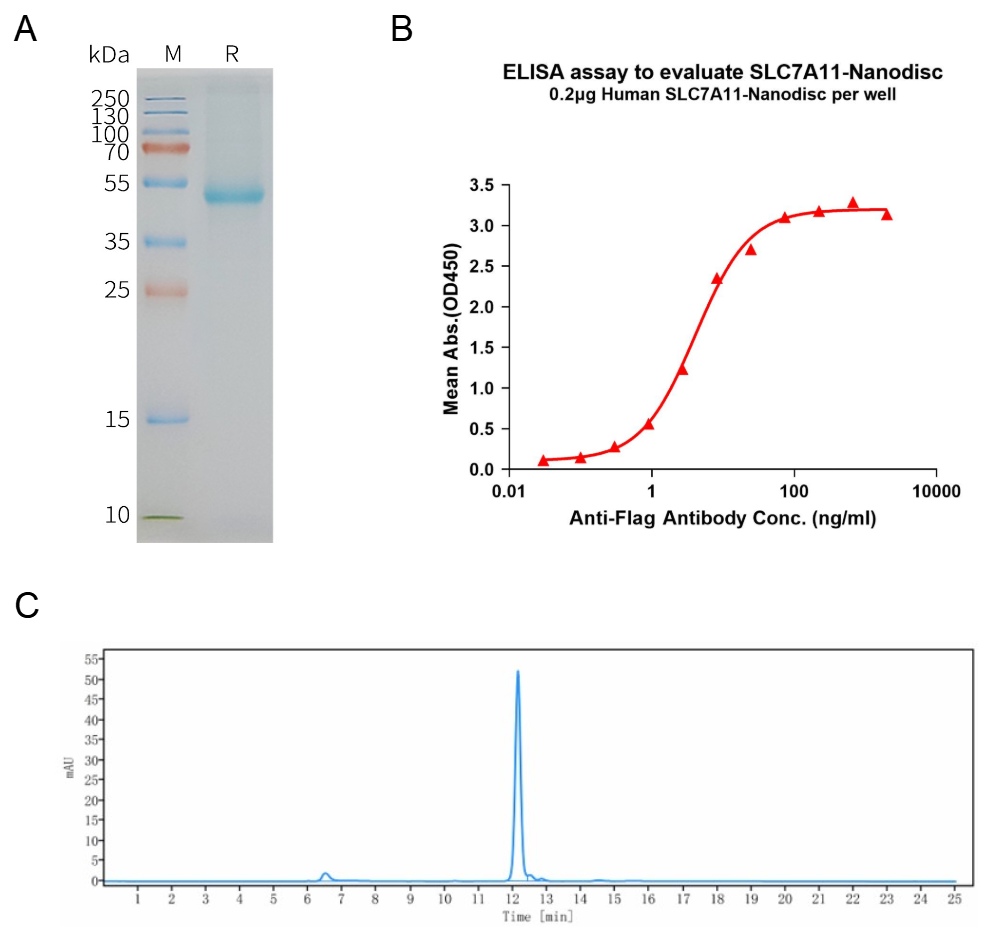
A: Human SLC7A11-Nanodisc, Flag Tag on SDS-PAGE;
B: ELISA analysis of SLC7A11-Nanodisc can bind with anti-Flag monoclonal antibody and the EC50 is 4.101ng/ml;
C: The purity of Human SLC7A11-Nanodisc is greater than 90% as determined by SEC-HPLC.
Reference:
- Chen, X., et al. (2025). “Ferroptosis 2025: Decade of discovery and the future of therapy.” Cell.
- Gan, B., et al. (2024). “The multifaceted role of SLC7A11 in cancer: from ferroptosis to disulfidptosis.” Nature Reviews Cancer.
- Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021 Aug;12(8):599-620.
- Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012.
- Jiang L, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015.
- Liu X, et al. Cystine transporter regulation of redox balance in cancer. Trends in Cancer. 2020.
- Liu, X., et al. (2023). “Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis.” Nature Cell Biology.
- Zheng, J., et al. (2024). “SLC7A11-mediated metabolism: a double-edged sword in cancer.” Cell Death & Disease.